Whether you’d like IDT to perform your on or off-target analysis for you or you simply have questions, our CRISPR experts are happy to help.
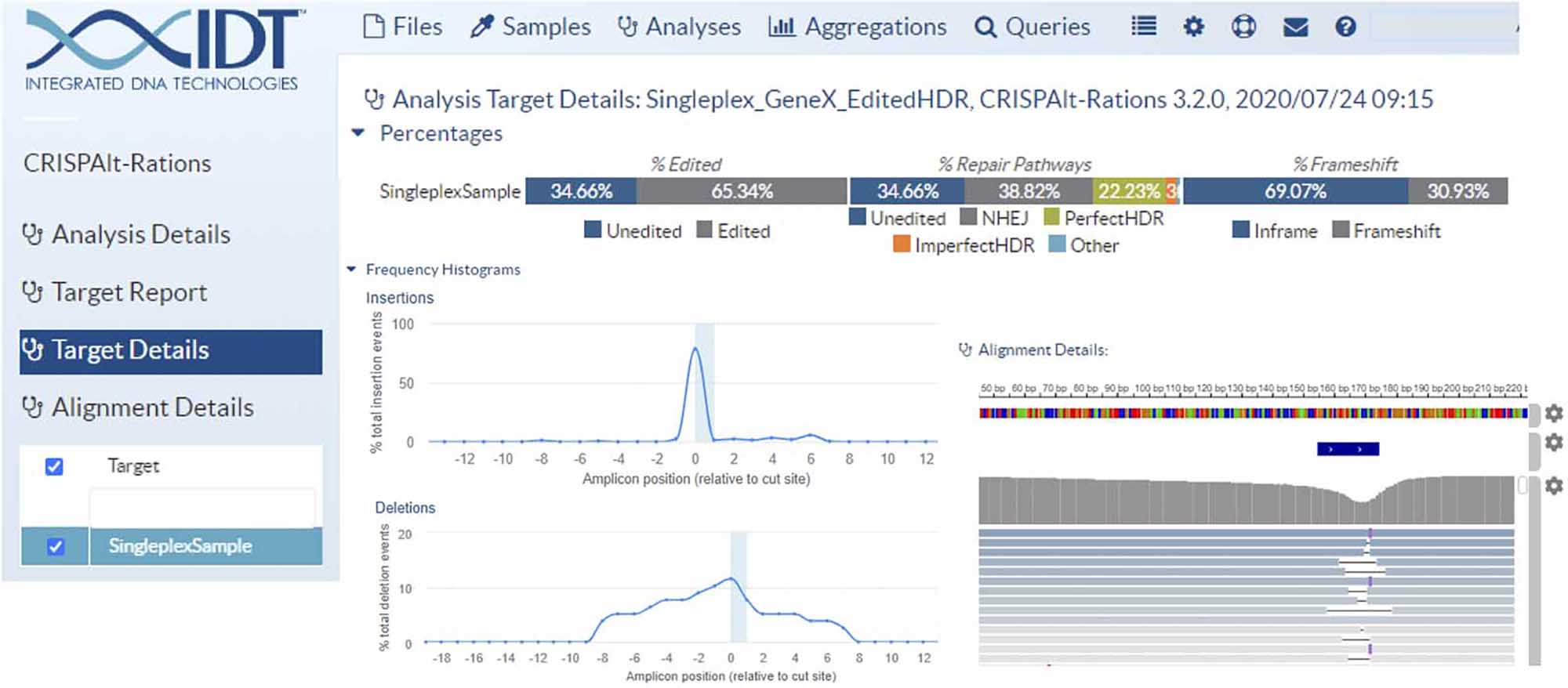
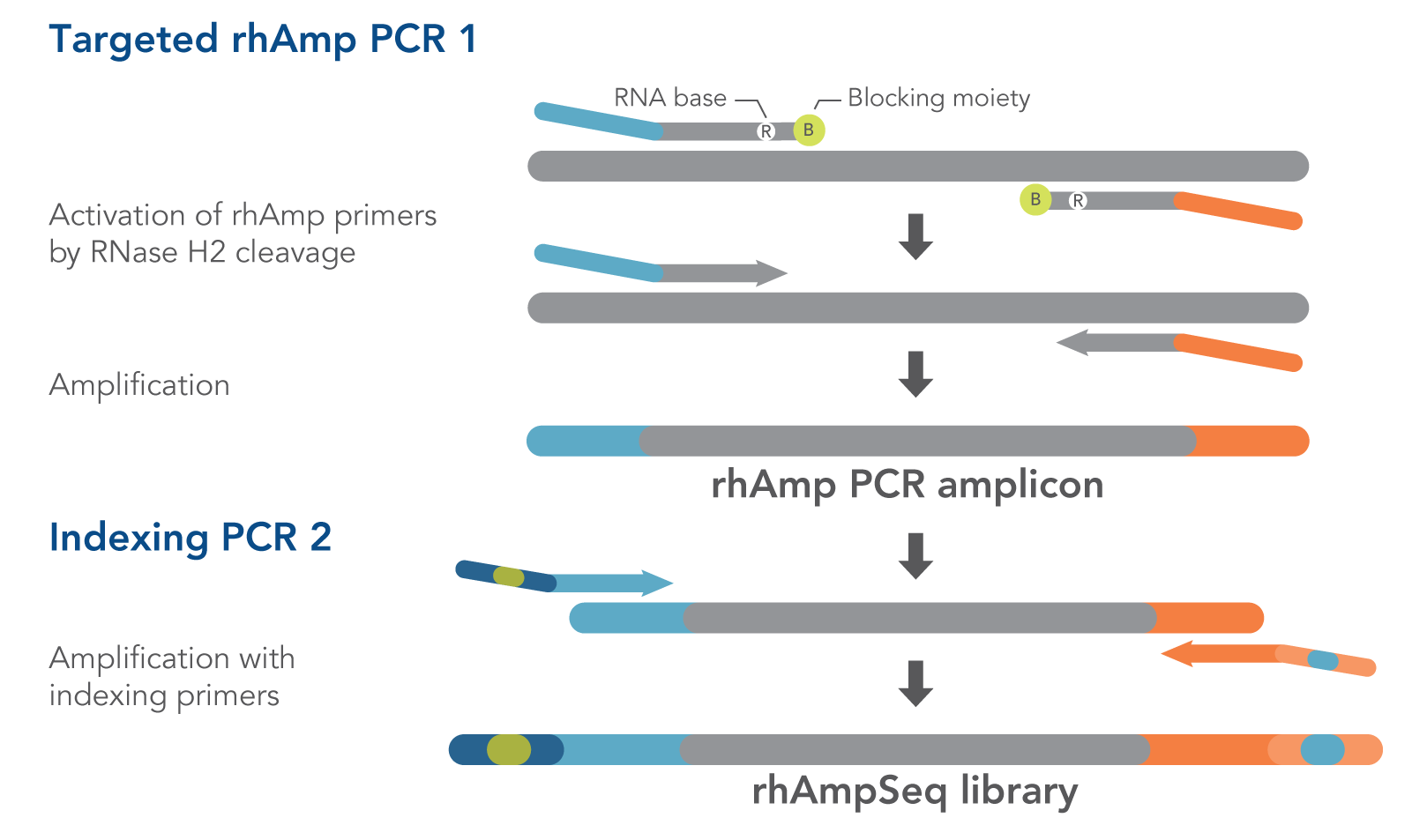
Quantify your editing results in under a week with three easy steps:
When your custom rhAmpSeq reagents arrive, follow the provided easy steps to build your rhAmpSeq multiplexed amplicon library, then sequence with Illumina-based NGS.
Upload your FASTQ files into the rhAmpSeq analysis portal for automated analysis of each edit site.
“IDT’s rhAmpSeq technology allows us to quickly and accurately quantify CRISPR genome editing off-target frequencies cost-effectively in our preclinical studies."

Dr. Ayal Hendel
Associate Professor
Bar-Ilan University
RUO26-4210_001
CGMP gRNA products are manufactured in accordance with ICH Q7, 21CFR210, 211, and parts of 600. IDT engineering runs and CGMP gRNA are for development and investigational use only. The performance characteristics of this product have not been established. This product is not intended to be used as final drug product. The purchaser is solely responsible for all decisions regarding the intended use of the product and any associated legal or regulatory obligations.